
FRUZAQLA 5 mg, gélule, boîte de 1 flacon de 21
Dernière révision : 26/11/2024
Taux de TVA : 2.1%
Prix de vente : 3 714,11 €
Taux remboursement SS : 100%
Base remboursement SS : 3 714,11 €
Laboratoire exploitant : TAKEDA FRANCE SAS
Source :

FRUZAQLA en monothérapie est indiqué pour le traitement des patients adultes atteints d'un cancer colorectal métastatique (CCRm) qui ont été traités antérieurement par les traitements standards disponibles, comprenant les chimiothérapies à base de fluoropyrimidine, d'oxaliplatine et d'irinotécan, les agents anti-VEGF et les agents anti-EGFR, et qui ont progressé ou sont intolérants au traitement par trifluridine-tipiracil ou par régorafénib.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Hypertension
Des cas d'hypertension, y compris des crises hypertensives, ont été rapportés chez des patients traités par fruquintinib (voir rubrique Effets indésirables). Chez les patients présentant une hypertension préexistante, celle-ci doit être surveillée et contrôlée de manière adéquate, conformément aux pratiques médicales standard, avant de commencer le traitement par fruquintinib.
L'hypertension doit faire l'objet d'une prise en charge médicale avec des médicaments antihypertenseurs et un ajustement de la posologie du fruquintinib, si nécessaire (voir rubrique Posologie et mode d'administration). Le fruquintinib doit être définitivement arrêté en cas d'hypertension ne pouvant être contrôlée par un traitement antihypertenseur ou chez les patients présentant une crise hypertensive.
Événements hémorragiques
Des événements hémorragiques ont été rapportés chez des patients traités par fruquintinib, y compris des événements au niveau du tractus gastro-intestinal (GI) (voir rubrique Effets indésirables). Des événements hémorragiques graves et parfois fatals ont été rapportés chez des patients après un traitement par fruquintinib.
Les profils hématologique et de coagulation doivent être surveillés conformément aux pratiques médicales standard chez les patients présentant un risque de saignement, y compris ceux traités par des anticoagulants ou d'autres médicaments concomitants qui augmentent le risque de saignement. En cas d'hémorragie sévère nécessitant une intervention médicale immédiate, le fruquintinib doit être définitivement arrêté (voir rubrique Posologie et mode d'administration).
Perforation gastro-intestinale
Des cas de perforation GI, dont des événements fatals, ont été rapportés chez des patients traités par fruquintinib (voir rubrique Effets indésirables).
Les symptômes de perforation GI doivent être régulièrement surveillés pendant le traitement par fruquintinib.
Le fruquintinib doit être définitivement arrêté chez les patients développant une perforation GI.
Protéinurie
Des cas de protéinurie sont survenus chez des patients traités par fruquintinib.
La protéinurie doit être surveillée avant l'instauration du traitement et pendant le traitement par fruquintinib, conformément aux pratiques médicales standard. En cas de détection d'une protéinurie ≥ 2 g/24 heures sur bandelette urinaire, il peut être nécessaire d'interrompre temporairement le traitement, d'adapter la posologie ou d'arrêter le traitement. Le fruquintinib doit être définitivement arrêté chez les patients développant un syndrome néphrotique (voir rubrique Posologie et mode d'administration).
Érythrodysesthésie palmo-plantaire (EPP)
L'EPP est l'effet indésirable dermatologique le plus fréquemment rapporté (voir rubrique Effets indésirables).
En cas de détection de réactions cutanées de grade ≥ 2, il peut être nécessaire d'interrompre temporairement le traitement, d'adapter la posologie ou d'arrêter le traitement (voir rubrique Posologie et mode d'administration).
Syndrome d'encéphalopathie postérieure réversible (SEPR)
Un cas de SEPR a été rapporté chez 1 patient (0,1 %) traité par fruquintinib dans les études cliniques (voir également rubrique Effets indésirables). Le SEPR est un trouble neurologique rare qui peut se manifester par des céphalées, des convulsions, une léthargie, une confusion, une altération des fonctions mentales, une cécité et d'autres troubles visuels ou neurologiques, avec ou sans hypertension associée. Le diagnostic de SEPR doit être confirmé par imagerie cérébrale, de préférence une imagerie par résonance magnétique (IRM). Chez les patients développant un SEPR, il est recommandé d'arrêter le fruquintinib, de contrôler l'hypertension et de prendre en charge les autres symptômes au moyen d'un traitement médical de support.
Altération de la cicatrisation des plaies
L'altération de la cicatrisation des plaies a été rapportée chez un patient (0,1 %) traité par fruquintinib.
Il est recommandé aux patients d'interrompre le fruquintinib pendant au moins 2 semaines avant une intervention chirurgicale. Il convient de ne pas reprendre le fruquintinib avant un délai minimum de 2 semaines suivant l'intervention chirurgicale, en fonction des indications cliniques, lorsque la cicatrisation de la plaie est jugée satisfaisante.
Événements thromboemboliques artériels et veineux
Il est recommandé d'éviter de commencer un traitement par fruquintinib chez les patients avec des antécédents d'événements thromboemboliques (y compris de thrombose veineuse profonde et d'embolie pulmonaire) au cours des 6 derniers mois ou avec des antécédents d'accident vasculaire cérébral et/ou d'accident ischémique transitoire au cours des 12 derniers mois. En cas de suspicion de thrombose artérielle, le fruquintinib devra être arrêté immédiatement.
Excipients
Les gélules de fruquintinib 1 mg contiennent de la tartrazine (E102) et du jaune soleil FCF (E110), pouvant provoquer des réactions allergiques.
Les gélules de fruquintinib 5 mg contiennent du rouge allura AC (E129), pouvant provoquer des réactions allergiques.
Résumé du profil de sécurité
Les effets indésirables les plus fréquents sont l'hypertension (49,3 %), l'anorexie (35,6 %), la protéinurie (35,5 %), l'EPP (34,6 %), l'hypothyroïdie (32,4 %), la dysphonie (28,6 %), la diarrhée (26,3 %) et l'asthénie (24,5 %).
Les effets indésirables de grade ≥ 3 les plus fréquents sont l'hypertension (19,1 %) et l'EPP (8,3 %).
Les effets indésirables graves les plus fréquents sont l'hémorragie gastro-intestinale (1,5 %), la pneumonie (1,5 %), l'hypertension (1,5 %) et la perforation gastro-intestinale (1,3 %).
La fréquence des arrêts de traitement en raison d'effets indésirables est de 7,6 %. L'effet indésirable le plus fréquent entraînant un arrêt de traitement est la protéinurie (1,6 %).
La fréquence des réductions de dose en raison d'effets indésirables est de 20,5 %. Les effets indésirables les plus fréquents entraînant une réduction de la dose sont l'EPP (6,4 %), l'hypertension (3,7 %) et la protéinurie (3,4 %).
Tableau résumant les effets indésirables
Les fréquences des effets indésirables sont basées sur les données compilées d'études cliniques menées auprès de 911 patients atteints d'un CCRm précédemment traité. Les patients ont reçu au moins 1 dose (5 mg) de fruquintinib en monothérapie (5 mg une fois par jour, 3 semaines de traitement/1 semaine d'interruption) pendant une durée médiane de 3,68 mois.
Les effets indésirables rapportés dans les études cliniques ou lors de l'utilisation du fruquintinib après sa mise sur le marché sont listés dans le tableau 3 par classe de systèmes d'organes MedDRA et par fréquence. Au sein de chaque classe de systèmes d'organes, les effets indésirables sont classés selon un ordre décroissant de fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données postcommercialisation disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 3 : Effets indésirables signalés chez les patients atteints d'un CCRm traités par fruquintinib (N = 911)
|
Classe de systèmes d'organes |
Catégorie de fréquence |
Effets indésirables Tous grades |
|
Infections et infestations |
Fréquent |
Pneumonie Infection des voies aériennes supérieures1 Infections bactériennes2 |
|
Affections hématologiques et du système lymphatique |
Très fréquent |
Thrombopénie3 |
|
Fréquent |
Leucopénie4 Neutropénie5 |
|
|
Affections endocriniennes |
Très fréquent |
Hypothyroïdie6 |
|
Troubles du métabolisme et de la nutrition |
Très fréquent |
Anorexie7 |
|
Fréquent |
Hypokaliémie |
|
|
Affections du système nerveux |
Peu fréquent |
Syndrome d'encéphalopathie postérieure réversible* |
|
Affections vasculaires |
Très fréquent |
Hypertension8 |
|
Fréquence indéterminée |
Dissection aortique† |
|
|
Affections respiratoires, thoraciques et médiastinales |
Très fréquent |
Dysphonie9 |
|
Fréquent |
Épistaxis Mal de gorge10 |
|
|
Affections gastro-intestinales |
Très fréquent |
Diarrhée Stomatite11 |
|
Fréquent |
Hémorragie gastro-intestinale12 Perforation gastro-intestinale13 Enzymes pancréatiques augmentées14 Douleur buccale15 |
|
|
Peu fréquent |
Pancréatite16 |
|
|
Affections hépatobiliaires |
Très fréquent |
Aspartate aminotransférase augmentée Bilirubine totale augmentée17 Alanine aminotransférase augmentée |
|
Peu fréquent |
Cholécystite18 |
|
|
Affections de la peau et du tissu souscutané |
Très fréquent |
Érythrodysesthésie palmo-plantaire |
|
Fréquent |
Rash19 |
|
|
Affections musculosquelettiques et du tissu conjonctif |
Très fréquent |
Gêne musculosquelettique20 Arthralgie |
|
Affections du rein et des voies urinaires |
Très fréquent |
Protéinurie21 |
|
Troubles généraux et anomalies au site d'administration |
Très fréquent |
Asthénie Fatigue |
|
Fréquent |
Inflammation muqueuse |
|
|
Peu fréquent |
Altération de la cicatrisation des plaies*,22 |
Les données de sécurité sont basées sur l'ensemble des patients atteints d'un CCRm qui ont reçu au moins 1 dose (5 mg) de fruquintinib en monothérapie (5 mg une fois par jour, 3 semaines de traitement/1 semaine d'interruption) dans les études groupées suivantes : 2012-013-00CH1 ; 2013-013-00CH1/FRESCO ; 2019-013-GLOB1/FRESCO-2, y compris la cohorte japonaise préliminaire en ouvert pour l'évaluation de la sécurité ; 2009-013-00CH1 ; 2012 013-00CH3 ; 2015-013-00US1. *Rapporté dans les études cliniques et dans le cadre du suivi post-commercialisation.
†Rapporté dans le cadre du suivi post-commercialisation.
Les termes suivants représentent un groupe d'événements liés qui décrivent un état pathologique plutôt qu'un événement isolé :
1L'infection des voies aériennes supérieures comprend la rhinopharyngite, la pharyngite et l'infection des voies aériennes supérieures.
2Les infections bactériennes comprennent la bactériurie asymptomatique, l'infection bactérienne, la bactériurie, la cellulite, la colite à Clostridium difficile, l'infection à Clostridium difficile, le sepsis à Enterobacter, l'infection des voies urinaires par colibacille, la folliculite, le furoncle, la paronychie, la pharyngite streptococcique, la bactériémie à streptocoque, l'infection bactérienne des voies urinaires, l'infection staphylococcique des voies urinaires.
3La thrombopénie comprend la numération plaquettaire diminuée, la thrombopénie.
4La leucopénie comprend la leucopénie, les globules blancs diminués.
5La neutropénie comprend la neutropénie, les neutrophiles diminués.
6L'hypothyroïdie comprend la TSH sanguine augmentée, l'hypothyroïdie.
7L'anorexie comprend l'appétit diminué, la perte de poids.
8L'hypertension comprend la pression artérielle diastolique augmentée, la pression artérielle augmentée, l'hypertension diastolique, l'hypertension, la crise hypertensive.
9La dysphonie comprend l'aphonie, la dysphonie.
10La douleur dans la gorge comprend la gêne laryngée, la douleur laryngée, la gêne oropharyngée, la douleur oropharyngée.
11La stomatite comprend l'ulcère aphteux, l'ulcération gingivale, l'ulcération buccale, la stomatite, l'ulcération linguale.
12L'hémorragie gastro-intestinale comprend l'hémorragie anale, l'hémorragie de l'anastomose, l'hémorragie gastrique, l'hémorragie gastro-intestinale, l'hématochézie, l'hémorragie hémorroïdaire, l'hémorragie intestinale, l'hémorragie gastrointestinale inférieure, l'hémorragie rectale, l'hémorragie gastro-intestinale supérieure.
13La perforation gastro-intestinale comprend la perforation gastrique, l'ulcère gastrique perforé, la perforation gastrointestinale, la perforation intestinale, la perforation du gros intestin, la perforation rectale, la perforation de l'intestin grêle.
14Les enzymes pancréatiques augmentées comprennent l'amylase augmentée, l'hyperamylasémie, l'hyperlipasémie, la lipase augmentée.
15La douleur buccale comprend la douleur gingivale, la douleur buccale, la douleur dentaire.
16La pancréatite comprend la pancréatite, la pancréatite aiguë.
17La bilirubine totale augmentée comprend la bilirubine conjuguée augmentée, la bilirubine sanguine augmentée, la bilirubine libre sanguine augmentée, l'hyperbilirubinémie, l'ictère, l'ictère cholestatique.
18La cholécystite comprend la cholécystite, la cholécystite aiguë, la cholécystite infectieuse.
19Le rash comprend le rash, le rash érythémateux, le rash maculeux, le rash maculopapuleux, le rash papuleux, le rash prurigineux.
20La gêne musculosquelettique comprend la douleur osseuse, les spasmes musculaires, la douleur musculosquelettique du thorax, la douleur musculosquelettique, la douleur cervicale, les extrémités douloureuses.
21La protéinurie comprend l'albuminurie, les protéines urinaires présentes, la protéinurie.
22L'altération de la cicatrisation des plaies comprend le retard de cicatrisation, le lâchage de suture.
Description de certains effets indésirables
Les données relatives aux effets indésirables sélectionnés suivants sont basées sur les patients ayant reçu au moins 1 dose (5 mg) de fruquintinib (5 mg une fois par jour, 3 semaines de traitement/1 semaine d'interruption) dans trois études randomisées contrôlées contre placebo (2012013-00CH1 ; 2013-013-00CH1/FRESCO ; 2019-013-GLOB1/FRESCO-2). Les recommandations de prise en charge de ces effets indésirables sont décrites à la rubrique Mises en garde spéciales et précautions d'emploi.
Hypertension
Une hypertension a été rapportée chez 47,4 % des patients du bras traité par le fruquintinib. Environ la moitié de ces événements sont survenus au cours des 2 premières semaines suivant l'instauration du traitement par fruquintinib. Des cas d'hypertension de grade ≥ 3 ont été rapportés chez 18,4 % des patients du bras traité par le fruquintinib. Le délai médian d'apparition d'une hypertension chez les patients traités par fruquintinib était de 15 jours (intervalle : 1 jour à 7,6 mois). Trois patients (0,4 %) traités par fruquintinib ont présenté une crise hypertensive. La majorité des événements ont régressé ou se sont résolus après une interruption du traitement ou une réduction de la dose, survenant respectivement chez 3,1 % et 3,7 % des patients. L'hypertension a entraîné un arrêt définitif du traitement chez 0,5 % des patients.
Événements hémorragiques
Des événements hémorragiques ont été rapportés chez 26,5 % des patients du bras recevant le fruquintinib et 14,6 % des patients du bras recevant le placebo. La plupart des événements hémorragiques survenus chez les patients traités par fruquintinib étaient de gravité légère à modérée (le taux d'incidence des événements hémorragiques de grade ≥ 3 était de 2,0 % dans le bras recevant le fruquintinib. Le délai médian d'apparition chez les patients traités par fruquintinib était de 23 jours (intervalle : 1 jour à 9,8 mois). Des événements hémorragiques fatals ont été rapportés chez 0,5 % des patients du groupe recevant le fruquintinib. Le taux d'incidence des événements hémorragiques ayant entraîné un arrêt du traitement était de 1,2 %. Les réactions hémorragiques les plus fréquentes étaient les hémorragies gastro-intestinales (7 %) et les épistaxis (5,6 %). L'événement hémorragique grave le plus fréquemment rapporté était l'hémorragie gastro-intestinale, qui a été signalée chez 1,5 % des patients du bras recevant le fruquintinib contre 0,5 % du bras recevant le placebo.
Perforation gastro-intestinale (GI)
Des cas de perforation gastro-intestinale ont été rapportés chez 1,5 % des patients du bras recevant le fruquintinib. Une perforation GI fatale a été rapportée chez 0,1 % des patients traités par fruquintinib. La perforation intestinale (0,8 %) constituait le type de perforation GI le plus fréquent. Le taux d'incidence des événements de perforation gastro-intestinale ayant entraîné un arrêt du traitement était de 1,0 %.
Protéinurie
Une protéinurie a été rapportée chez 32,9 % des patients du bras recevant le fruquintinib. La plupart des événements de protéinurie survenus chez les patients traités par fruquintinib étaient de gravité légère à modérée (le taux d'incidence des événements de protéinurie de grade ≥ 3 était de 2,8 % dans le bras recevant le fruquintinib. Le délai médian d'apparition d'une protéinurie chez les patients traités par fruquintinib était de 28 jours (intervalle : 6 jours à 1,3 an). La plupart des événements ont régressé ou se sont résolus après une interruption du traitement ou une réduction de la dose. La protéinurie a entraîné un arrêt définitif du traitement chez 1,8% des patients traités par fruquintinib.
Érythrodysesthésie palmo-plantaire (EPP)
Une érythrodysesthésie palmo-plantaire a été rapportée chez 32,7 % des patients du bras recevant le fruquintinib. Le taux d'incidence des EPP de grade ≥ 3 dans le bras recevant le fruquintinib était de 8,5 %. Le délai médian d'apparition d'une EPP chez les patients traités par fruquintinib était de 20 jours (intervalle : 1 jour à 7,4 mois). La majorité des événements ont régressé ou se sont résolus après une interruption du traitement ou une réduction de la dose, survenant respectivement chez 6,4 % et 6,3 % des patients. L'EPP a entraîné un arrêt définitif du traitement chez 0,5 % des patients.
Syndrome d'encéphalopathie postérieure réversible (SEPR)
Un cas (0,1 %) de SEPR (grade 4) a été rapporté chez les patients ayant reçu le fruquintinib en monothérapie au cours des études cliniques. Des cas de SEPR ont également été rapportés au cours du suivi post-commercialisation. Tous les cas de SEPR ont été résolus après un arrêt du traitement.
Hypothyroïdie
Une hypothyroïdie a été rapportée chez 31,5 % des patients du bras recevant le fruquintinib. Le taux d'incidence du dysfonctionnement thyroïdien de grade ≥ 3 dans le bras recevant le fruquintinib était faible (0,3 %). Le délai médian d'apparition d'une hypothyroïdie chez les patients traités par fruquintinib était de 56 jours (intervalle : 18 jours à 1,4 an). Aucun événement n'a entraîné de réduction de la dose ou d'arrêt du traitement.
Infections
Des infections ont été rapportées chez 23,4 % des patients du groupe recevant le fruquintinib et 13,3 % des patients du groupe recevant le placebo. La plupart des événements infectieux survenus chez les patients traités par fruquintinib étaient de gravité légère à modérée (le taux d'incidence des infections de grade ≥ 3 était de 6 % dans le bras recevant le fruquintinib. Des infections graves ont été signalées chez 4,1 % des patients du bras recevant le fruquintinib et des événements infectieux fatals ont été signalés chez 1,0 % des patients du bras recevant le fruquintinib. Le taux d'incidence des infections ayant entraîné un arrêt du traitement était de 0,9 %. La réaction infectieuse la plus fréquente était l'infection des voies respiratoires supérieures (5,0 %). L'infection grave la plus fréquemment rapportée était la pneumonie (1,4 %).
Anomalies du bilan hépatique
Des anomalies du bilan hépatique ont été rapportées chez 36,4 % des patients du bras recevant le fruquintinib et 23,5 % des patients du groupe recevant le placebo. La plupart des affections hépatobiliaires survenues chez les patients traités par fruquintinib étaient de gravité légère à modérée (le taux d'incidence des anomalies du bilan hépatique de grade ≥ 3 était de 8,8 % dans le bras recevant le fruquintinib. Les événements d'anomalie du bilan hépatique les plus fréquents étaient l'augmentation du taux d'ASAT (18,1 %), l'augmentation du taux de bilirubine totale (18,3 %) et l'augmentation du taux d'ALAT (15,5 %). Le délai médian d'apparition d'une anomalie du bilan hépatique chez les patients traités par fruquintinib était de 28 jours (intervalle : 4 jours à 12 mois). Des anomalies du bilan hépatique graves ont été signalées chez 2,3 % des patients et des anomalies du bilan hépatique fatales ont été signalées chez 0,3 % des patients du bras recevant le fruquintinib. Des anomalies du bilan hépatique ont entraîné une interruption du traitement et une réduction de la dose chez respectivement 4,6 % et 2,0 % des patients, et un arrêt définitif du traitement chez 1,5 % des patients.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration – voir Annexe V.
SURVEILLANCE du traitement :
- Symptômes de perforation gastro-intestinale, régulièrement pendant le traitement.
- Protéinurie, avant l'instauration du traitement et pendant le traitement.
RECOMMANDER aux femmes en âge de procréer d'utiliser une contraception
effective pendant le traitement et jusqu'à au moins 2 semaines après la
dernière dose de fruquintinib.
Femmes en âge de procréer/Contraception chez la femme
Il convient de recommander aux femmes en âge de procréer d'utiliser une contraception effective pendant le traitement et jusqu'à au moins 2 semaines après la dernière dose de fruquintinib.
Grossesse
Il n'existe pas de données cliniques disponibles sur l'utilisation du fruquintinib chez la femme enceinte.
Sur la base de son mécanisme d'action, le fruquintinib est susceptible d'être nocif pour le fœtus. Les études effectuées chez l'animal ont mis en évidence une toxicité pour la reproduction, notamment des malformations fœtales (voir rubrique Données de sécurité préclinique). FRUZAQLA ne doit pas être utilisé pendant la grossesse, sauf si l'état clinique de la femme nécessite un traitement par fruquintinib.
Si le fruquintinib est utilisé pendant la grossesse ou si la patiente débute une grossesse pendant le traitement, la patiente doit être informée du risque potentiel pour le fœtus.
Allaitement
La sécurité d'emploi du fruquintinib pendant l'allaitement n'a pas été établie. Il est inconnu si le fruquintinib ou ses métabolites sont excrétés dans le lait maternel. Il n'existe pas de données animales sur l'excrétion du fruquintinib dans le lait animal. Un risque pour les nouveau-nés/nourrissons allaités ne peut être exclu.
L'allaitement doit être interrompu au cours du traitement et pendant 2 semaines après la dernière dose.
Fertilité
Il n'existe pas de données concernant les effets du fruquintinib sur la fertilité chez l'être humain. Les résultats des études effectuées chez l'animal indiquent que le fruquintinib peut altérer la fertilité des mâles et des femelles (voir rubrique Données de sécurité préclinique).
Effets d'autres médicaments sur la pharmacocinétique du fruquintinib
Inducteurs du CYP3A
L'administration concomitante de fruquintinib et de rifampicine (un puissant inducteur du CYP3A) à une dose de 600 mg une fois par jour a entraîné une diminution de 65 % de l'ASCinf du fruquintinib et a entraîné une diminution de 12 % de la Cmax. Il convient d'éviter l'utilisation concomitante du fruquintinib avec des inducteurs puissants et modérés du CYP3A.
Inhibiteurs du CYP3A
L'administration concomitante de fruquintinib et d'itraconazole (un puissant inhibiteur du CYP3A) à une dose de 200 mg deux fois par jour n'a pas entraîné de modifications cliniquement significatives de l'aire sous la courbe de concentration en fonction du temps (ASC) et de la Cmax du fruquintinib. Aucune adaptation posologique de fruquintinib n'est nécessaire en cas d'utilisation concomitante avec des inhibiteurs du CYP3A.
Antiacides
L'administration concomitante de fruquintinib et de rabéprazole (un inhibiteur de la pompe à protons) à une dose de 40 mg une fois par jour n'a pas entraîné de modifications cliniquement significatives de l'ASC du fruquintinib. Aucune adaptation posologique de fruquintinib n'est nécessaire en cas d'utilisation concomitante avec des antiacides.
Effet du fruquintinib sur la pharmacocinétique d'autres médicaments
Médicaments substrats de la glycoprotéine P (P-gp)
L'administration concomitante d'une dose unique de 150 mg de dabigatran étexilate (un substrat de la P-gp) et d'une dose unique de 5 mg de fruquintinib a entraîné une diminution de 9 % l'ASC du dabigatran. Aucune adaptation posologique n'est recommandée pour les substrats de la P-gp en cas d'utilisation concomitante avec le fruquintinib.
Médicaments substrats de la protéine de résistance au cancer du sein (BCRP)
L'administration concomitante d'une dose unique de 10 mg de rosuvastatine (un substrat de la BCRP) et d'une dose unique de 5 mg de fruquintinib a entraîné une diminution de 19 % de l'ASC de la rosuvastatine. Aucune adaptation posologique n'est recommandée pour les substrats de la BCRP en cas d'utilisation concomitante avec le fruquintinib.
FRUZAQLA doit être instauré par un médecin ayant l'expérience de l'administration des traitements anticancéreux.
Posologie
La dose recommandée de fruquintinib est de 5 mg (une gélule de 5 mg) une fois par jour, approximativement à la même heure, pendant 21 jours consécutifs, suivis d'une période sans traitement de 7 jours, constituant un cycle complet de 28 jours.
Durée du traitement
Le traitement par fruquintinib doit être poursuivi jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable.
Oubli de doses ou vomissements
Si l'oubli de la dose remonte à moins de 12 heures, la dose oubliée doit être prise et la dose suivante doit être prise au moment prévu.
Si l'oubli de la dose remonte à plus de 12 heures, la dose oubliée ne doit pas être prise et la dose suivante doit être prise au moment prévu.
Si un patient vomit après avoir pris une dose, il ne doit pas reprendre la dose le même jour, mais suivre la posologie habituelle telle que prescrite le jour suivant.
Adaptations posologiques en cas d'effets indésirables
La dose doit être modifiée en fonction de la sécurité et de la tolérance. Le fruquintinib doit être arrêté définitivement chez les patients ne tolérant pas une dose de 3 mg une fois par jour. Le schéma de réduction recommandée de la dose en cas d'effets indésirables est présenté dans le tableau 1.
Tableau 1 : Schéma de réduction recommandée de la posologie de FRUZAQLA
|
Schéma de réduction de la dose |
Posologie |
Nombre et dosage des gélules |
|
Première réduction de la dose |
4 mg une fois par jour |
Quatre gélules de 1 mg une fois par jour |
|
Deuxième réduction de la dose |
3 mg une fois par jour |
Trois gélules de 1 mg une fois par jour |
Les modifications recommandées de la dose en cas d'effets indésirables sont présentées dans le tableau 2.
Tableau 2 : Modification recommandée de la dose de FRUZAQLA en cas d'effets indésirables
|
Effet indésirable |
Sévérité1 |
Modification de la dose |
|
Hypertension |
Grade 3 |
• Interrompre temporairement le traitement en cas de persistance de l'hypertension de grade 3 malgré l'instauration ou la modification d'un traitement antihypertenseur. • En cas de retour de l'hypertension à un grade 1 ou à la valeur de référence, reprendre le traitement à une dose réduite conformément au tableau 1.
Si le patient présente encore une hypertension de grade 3 après avoir pris une dose de 3 mg par jour, arrêter définitivement le traitement. |
|
Grade 4 |
Arrêter définitivement le traitement. |
|
|
Événements hémorragiques |
Grade 2 |
• Interrompre temporairement le traitement jusqu'à ce que l'hémorragie se résorbe complètement ou revienne à un grade 1. • Reprendre le traitement à une dose réduite conformément au tableau 1.
Si le patient présente encore des événements hémorragiques de grade 2 après avoir pris une dose de 3 mg par jour, arrêter définitivement le traitement. |
|
Grade ≥ 3 |
Arrêter définitivement le traitement. |
|
|
Protéinurie |
≥ 2 g / 24 heures |
• Interrompre temporairement le traitement jusqu' à ce que la protéinurie se résorbe complètement ou soit < 1 g / 24 heures ou à la valeur de référence. • Reprendre le traitement à une dose réduite conformément au tableau 1.
Si le patient présente encore une protéinurie ≥ 2 g/24 heures après avoir pris une dose de 3 mg par jour, arrêter définitivement le traitement.
Arrêter définitivement le traitement en cas de syndrome néphrotique. |
|
Anomalies des tests de la fonction hépatique |
Anomalies des tests de la fonction hépatique de grade 2 ou 3. |
• Interrompre temporairement le traitement jusqu'à ce que l'anomalie du bilan hépatique revienne au grade 1 ou aux valeurs de référence. • Reprendre le traitement à une dose réduite conformément au tableau 1.
Si le patient présente encore des anomalies du bilan hépatique de grade 2 ou 3 après avoir pris une dose de 3 mg par jour, arrêter définitivement le traitement. |
|
Élévation de grade ≥ 2 (> 3 x LSN) du taux d'ALAT ou d'ASAT avec élévation de la bilirubine totale concomitante > 2 × LSN en l'absence de cholestase ; anomalies du bilan hépatique de grade 4. |
Arrêter définitivement le traitement. |
|
|
Érythrodysesthésie palmo-plantaire (EPP) |
Grade 2 |
• Administrer un traitement de support. • Interrompre temporairement le traitement jusqu'au retour de l'EPP à un grade 1 ou à la valeur de référence. • Reprendre le traitement au même palier de dose. |
|
Grade 3 |
• Administrer un traitement de support. • Interrompre temporairement le traitement jusqu'au retour de l'EPP à un grade 1 ou à la valeur de référence. • Reprendre le traitement à une dose réduite conformément au tableau 1.
Si le patient présente encore de l'EPP de Grade 3 après avoir pris une dose de 3 mg par jour, arrêter définitivement le traitement. |
|
|
Autres effets indésirables |
Grade 3 |
• Interrompre temporairement le traitement jusqu'au retour de l'effet indésirable à un grade 1 ou à la valeur de référence. • Reprendre le traitement à une dose réduite conformément au tableau 1.
Si le patient présente encore d'autres effets indésirables de Grade 3 après avoir pris une dose de 3 mg par jour, arrêter définitivement le traitement. |
|
Grade 4 |
Arrêter le traitement.
Envisager de reprendre le traitement à une dose réduite conformément au tableau 1 en cas de retour de la toxicité à un grade 1 ou à la valeur de référence et si le bénéfice potentiel l'emporte sur les risques. |
1 Les grades de toxicité correspondent à la classification du ‘National Cancer Institute Common Terminology Criteria for Adverse Events.' version 5.0 (NCI-CTCAE v5).
Populations particulières
Insuffisance rénale
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance rénale légère, modérée ou sévère (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée (voir rubrique Propriétés pharmacocinétiques).
FRUZAQLA n'est pas recommandé chez les patients présentant une insuffisance hépatique sévère, car la sécurité et l'efficacité de FRUZAQLA n'ont pas été étudiées dans cette population.
Personne âgée
Aucune adaptation posologique n'est nécessaire chez les patients âgés de 65 ans ou plus.
Population pédiatrique
Il n'existe pas d'utilisation justifiée de FRUZAQLA dans la population pédiatrique dans l'indication de cancer colorectal métastatique.
Mode d'administration
FRUZAQLA est destiné à une administration par voie orale.
Les gélules peuvent être prises pendant ou en dehors des repas et doivent être avalées entières.
Les gélules ne doivent pas être mâchées, dissoutes, ni ouvertes, car on ne connaît pas les effets potentiels de ces altérations.
Durée de conservation :
2 ans.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation concernant la température.
À conserver dans l'emballage d'origine à l'abri de l'humidité. Conserver le flacon soigneusement fermé.
Sans objet.
Dans les études cliniques, la plus forte dose de fruquintinib étudiée était de 6 mg par jour.
Les effets d'un surdosage de fruquintinib sont inconnus, et il n'existe pas d'antidote connu en cas de surdosage de fruquintinib. En cas de surdosage, le fruquintinib doit être interrompu et des mesures générales symptomatiques ainsi qu'une surveillance jusqu'à stabilisation clinique doivent être mises en œuvre.
Classe pharmacothérapeutique : Agents antinéoplasiques, inhibiteurs de la tyrosine kinase du récepteur du facteur de croissance de l'endothélium vasculaire (VEGFR), Code ATC : L01EK04
Mécanisme d'action et effets pharmacodynamiques
Le fruquintinib est un inhibiteur sélectif de la tyrosine kinase des VEGFR-1, -2 et -3, dont les effets antitumoraux résultent de la suppression de l'angiogenèse tumorale.
Électrophysiologie cardiaque
Aucune prolongation de l'intervalle QT corrigé en fonction de la fréquence cardiaque (QTc) (> 10 millisecondes) n'a été observée à la posologie recommandée de fruquintinib. Une analyse concentration-QT (N = 205) n'a pas mis en évidence d'association entre les concentrations plasmatiques de fruquintinib et les modifications de l'intervalle QTc par rapport aux valeurs au début du traitement.
Efficacité et sécurité cliniques
L'efficacité et la sécurité du fruquintinib associé aux meilleurs soins de support (‘best supportive care' (BSC)) ont été évaluées dans une étude de phase III randomisée, contrôlée contre placebo, en double aveugle (étude FRESCO-2) menée chez des patients atteints d'un CCRm précédemment traités, notamment par des chimiothérapies à base d'oxaliplatine ou d'irinotécan. L'efficacité clinique du fruquintinib dans l'étude FRESCO-2 est décrite ci-après.
Étude FRESCO-2
L'efficacité clinique et la sécurité du fruquintinib ont fait l'objet d'une évaluation dans le cadre d'une étude de phase III multicentrique, internationale, randomisée, en double aveugle, contrôlée contre placebo (étude FRESCO-2), menée chez 691 patients atteints d'un CCRm précédemment traités par des traitements standard approuvés - notamment par chimiothérapie à base de fluoropyrimidine, d'oxaliplatine et d'irinotécan, par un traitement biologique anti-VEGF, un traitement anti-EGFR en cas de gène RAS de type sauvage, et dont la maladie a progressé sous trifluridine/tipiracil et/ou régorafenib ou qui ont présenté une intolérance à ces traitements. Les patients ont été considérés comme intolérants à la trifluridine/tipiracil ou au régorafenib s'ils ont reçu au moins 1 dose de l'un ou l'autre agent et que le traitement a été arrêté pour des raisons autres qu'une progression de la maladie. Les patients atteints de tumeurs MSI-H ou dMMR ont été précédemment traités par des inhibiteurs de point de contrôle immunitaire, et les patients atteints de tumeurs avec mutation V600E de BRAF ont été précédemment traités par un inhibiteur de BRAF, si le traitement était approuvé et disponible dans le pays ou la région du patient. La randomisation était stratifiée en fonction du traitement antérieur (trifluridine/tipiracil ou régorafenib ou à la fois trifluridine/tipiracil et régorafenib), du statut du gène RAS (type sauvage ou mutant) et de la durée de la maladie métastatique (≤ 18 mois ou > 18 mois).
Les patients ayant un indice de performance ECOG (‘Eastern Cooperative Oncology Group') ≥ 2, une fraction d'éjection ventriculaire gauche ≤ 50 %, une pression artérielle systolique > 140 mmHg ou une pression artérielle diastolique > 90 mmHg, des protéines urinaires ≥ 1 g/24 h ou un poids corporel < 40 kg ont été exclus. Le critère principal d'évaluation de l'efficacité était la survie globale (SG). Le principal critère secondaire d'évaluation de l'efficacité était la survie sans progression (SSP ; évaluée par l'investigateur à l'aide des critères d'évaluation de la réponse dans les tumeurs solides ‘Response Evaluation Criteria in Solid Tumours' [RECIST], version 1.1) et d'autres critères d'évaluation secondaires complémentaires comprenaient le taux de contrôle de la maladie.
Au total, 691 patients ont été randomisés (selon un rapport 2:1) pour recevoir 5 mg de fruquintinib par voie orale une fois par jour (N = 461) en association avec des soins de support ou un placebo par voie orale une fois par jour (N = 230) en association avec des soins de support (ci-après dénommés respectivement fruquintinib et placebo), pendant 21 jours de traitement suivis de 7 jours d'interruption du traitement dans le cadre d'un cycle de traitement de 28 jours.
Parmi les 691 patients randomisés, l'âge médian était de 64 ans (intervalle : 25 à 86 ans), 47 % des patients étant âgés de 65 ans ou plus. 55,7 % des patients étaient des hommes, 80,9 % étaient caucasiens et ils avaient un IP ECOG de 0 (43,1 %) ou 1 (56,9 %). Une tumeur de type RAS sauvage était signalé dans les tumeurs de 36,9 % des patients lors de l'entrée dans l'étude. La durée médiane de la maladie métastatique était de 39 mois (intervalle : 6 mois à 16,1 ans). Le nombre médian de lignes de traitement antérieures pour la maladie métastatique était de 4 (intervalle : 2 à 16).
En plus du traitement par chimiothérapie à base de fluoropyrimidine, d'oxaliplatine et d'irinotécan, 96,4 % des patients avaient déjà reçu un traitement anti-VEGF, 38,8 % un traitement anti-EGFR, 52,2 % une association trifluridine/tipiracil et 8,4 % du régorafenib, et 39,4 % avaient déjà reçu à la fois une association trifluridine/tipiracil et du régorafenib, 4,6 % avaient déjà reçu une immunothérapie et 2,3 % un inhibiteur de BRAF.
Dans l'étude FRESCO-2, l'ajout du fruquintinib aux meilleurs soins de support a entraîné une amélioration statistiquement significative de la SG et de la SSP par rapport au placebo associé aux meilleurs soins de support (voir tableau 4 et figure 1).
Tableau 4 : Résultats d'efficacité de l'étude FRESCO-2
|
Critère d'évaluation |
Fruquintinib (N = 461) |
|
Placebo (N = 230) |
|
SG |
|
|
|
|
Médiane en mois (IC à 95 %) |
7,4 (6,7 ; 8,2) |
|
4,8 (4,0 ; 5,8) |
|
Hazard Ratio1 (IC à 95 %) |
|
0,66 (0,55 ; 0,80) |
|
|
Valeur p2 |
|
< 0,001 |
|
|
SSP3 |
|
|
|
|
Médiane en mois (IC à 95 %) |
3,7 (3,5 ; 3,8) |
|
1,8 (1,8 ; 1,9) |
|
Hazard ratio1 (IC à 95 %) |
|
0,32 (0,27 à 0,39) |
|
|
Valeur p2 |
|
< 0,001 |
|
Abréviations : IC : intervalle de confiance ; N = nombre de patients ; HR : hazard ratio ; SG : survie globale ; SSP : survie sans progression
La médiane de la SG et de la SSP a été calculée à l'aide de la méthode de Kaplan-Meier.
1Le HR et son IC à 95 % ont été estimés à l’aide du modèle de risques proportionnels de Cox stratifié (tenant
compte des facteurs de stratification), le groupe de traitement constituant la seule covariable du modèle.
2La valeur p (bilatérale) a été calculée à l’aide d’un test du log-rank stratifié pour tenir compte des facteurs de
stratification.
3Évalué par l’investigateur à l’aide des critères RECIST, version 1.1.
Figure 1 : Courbe de Kaplan-Meier pour la survie globale dans l'étude FRESCO-2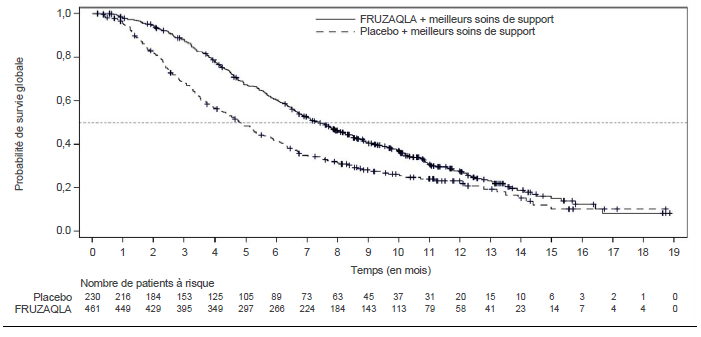
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec FRUZAQLA dans tous les sous-groupes de la population pédiatrique dans l'indication du cancer colorectal métastatique (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Absorption
Après l'administration orale de fruquintinib, le délai médian pour atteindre la concentration plasmatique maximale de fruquintinib (Tmax) était d'environ 2 heures. Le fruquintinib a montré un second pic d'absorption environ 24 heures après l'administration du médicament. Après l'administration répétée d'une dose quotidienne, l'exposition au fruquintinib (Cmax et ASC0-24h) a augmenté de manière proportionnelle à la dose dans l'intervalle de doses de 1 à 6 mg (0,2 à 1,2 fois la dose recommandée). Après l'administration de 5 mg de fruquintinib une fois par jour pendant 21 jours avec 7 jours d'interruption du traitement dans chaque cycle de 28 jours chez des patients atteints de tumeurs solides avancées, l'état d'équilibre du fruquintinib a été atteint après 14 jours et l'accumulation moyenne basée sur l'ASC0-24h a été multipliée par 4 par rapport à une dose unique. À la dose recommandée de 5 mg de fruquintinib, la moyenne géométrique (% de coefficient de variation (CV)) de la Cmaxet de l'ASC0-24h du fruquintinib à l'état d'équilibre était respectivement de 300 ng/mL (28 %) et de 5 880 ng*h/mL (29 %).
Effet de l'alimentation
Par rapport à une administration à jeun, un repas riche en matières grasses n'a pas eu d'effet cliniquement significatif sur la pharmacocinétique du fruquintinib chez des sujets sains. Le fruquintinib peut être administré au cours ou en dehors des repas.
Distribution
Le volume apparent de distribution du fruquintinib est d'environ 48,5 L. In vitro, la liaison du fruquintinib aux protéines plasmatiques est d'environ 95 % ; le fruquintinib se lie principalement à l'albumine sérique humaine.
Biotransformation
Le fruquintinib est métabolisé par plusieurs enzymes, dont le CYP450 (sous-familles CYP3A et CYP2C) et des systèmes enzymatiques non-CYP450. L'étude in vivo du métabolisme et du bilan de masse du fruquintinib marqué au [14C] a montré que le fruquintinib existe principalement dans le plasma humain sous sa forme inchangée, représentant environ 72 % de l'exposition plasmatique totale, et que le métabolite N-déméthyle du fruquintinib, médié par le CYP3A4, représente environ 17 % de l'exposition plasmatique totale. Les autres voies métaboliques comprennent la mono-oxydation multisite, la O-déméthylation, la N-déméthylation, la modification du noyau O-quinazoline et l'hydrolyse des amides. Les métabolites de phase II sont principalement des conjugués d'acide glucuronique et d'acide sulfurique des produits de phase I.
Études in vitro
Enzymes cytochromes P450 : Le CYP3A4 était la principale enzyme parmi les isoformes du CYP impliquées dans le métabolisme du fruquintinib, avec des contributions mineures du CYP2C8, du CYP2C9 et du CYP2C19.À des concentrations pertinentes sur le plan thérapeutique, le fruquintinib n'est pas un inhibiteur des CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 et CYP3A, ni un inducteur des CYP1A2, CYP2B6, CYP3A.
Systèmes de transporteurs : Le fruquintinib n'est pas un substrat de la glycoprotéine P (P-gp), de la protéine de transport des anions organiques (OATP)1B1 ou de l'OATP1B3. Le fruquintinib a inhibé la glycoprotéine P (P-gp) et la protéine de résistance au cancer du sein (BCRP) de manière dosedépendante in vitro et a démontré une solubilité aqueuse pH-dépendante. À des concentrations pertinentes sur le plan thérapeutique, le fruquintinib n'est pas un inhibiteur de l'OATP1B1, de l'OATP1B3, du transporteur d'anions organiques (OAT)1, de l'OAT3, du transporteur de cations organiques (OCT)2, de la protéine d'extrusion multidrogue et toxine (MATE)1, ou de la MATE2-K.
Élimination
Chez des patients atteints de tumeurs solides avancées, la clairance apparente (CL/F) du fruquintinib est de 14,8 mL/min à l'état d'équilibre après une prise quotidienne. La demi-vie d'élimination moyenne du fruquintinib est d'environ 42 heures.
Après l'administration d'une dose unique de 5 mg de fruquintinib radiomarqué à des sujets sains, environ 60 % de la dose a été retrouvée dans les urines (0,5 % de la dose de fruquintinib sous forme inchangée) et 30 % de la dose a été retrouvée dans les fèces (5 % de la dose de fruquintinib sous forme inchangée).
Populations particulières
Insuffisance rénale
Sur la base des analyses pharmacocinétiques de population, une insuffisance rénale légère à modérée (clairance de la créatinine [ClCr] comprise entre 30 et 89 mL/min) n'a pas eu d'effet cliniquement significatif sur la pharmacocinétique du fruquintinib. Dans une étude pharmacocinétique, l'ASC0-inf et la Cmax du fruquintinib non lié étaient comparables chez les patients atteints d'insuffisance rénale modérée (ClCr 30 - 59 mL/min, N = 8) ou sévère (ClCr 15 - 29 mL/min, N = 8) par rapport aux patients présentant une fonction rénale normale (ClCr ≥ 90 mL/min, N = 8).
Insuffisance hépatique
Sur la base d'analyses pharmacocinétiques de population, aucune différence cliniquement significative n'a été observée au niveau de la pharmacocinétique du fruquintinib entre les patients présentant une fonction hépatique normale et ceux présentant une insuffisance hépatique légère (bilirubine totale ≤ LSN avec un taux d'ASAT supérieur à la LSN ou bilirubine totale > 1 à 1,5 fois la LSN quel que soit le taux d'ASAT). Sur la base d'une étude pharmacocinétique dédiée à l'insuffisance hépatique, après l'administration d'une dose orale unique de 2 mg de fruquintinib, aucune différence cliniquement significative n'a été observée au niveau de l'ASC du fruquintinib normalisée en fonction de la dose entre les patients présentant une insuffisance hépatique modérée (classe B selon le score de Child Pugh) et les sujets ayant une fonction hépatique normale.
Âge, poids corporel, sexe ou origine ethnique
Les analyses pharmacocinétiques de population ont montré que l'âge (18 à 82 ans), le poids corporel (48 à 108 kg), le sexe ou l'origine ethnique n'avaient pas d'effet cliniquement significatif sur la pharmacocinétique du fruquintinib.
Population pédiatrique
Aucune étude pharmacocinétique n'a été réalisée avec le fruquintinib chez des patients âgés de moins de 18 ans.
Le fruquintinib a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. Une fatigue peut survenir après la prise de fruquintinib (voir rubrique Effets indésirables).
Des études de toxicité par administration répétée et de toxicité pour la reproduction ont mis en évidence une toxicité du fruquintinib à des concentrations plasmatiques moyennes inférieures aux concentrations thérapeutiques attendues chez l'homme.
Toxicité par administration répétée
Dans des études de toxicité par administration répétée chez l'animal, des effets ont été identifiés sur les principaux organes cibles au niveau du tractus gastro-intestinal, du système hépatobiliaire, du système immunitaire, du système squelettique (fémur et dents), des reins, du système hématopoïétique et de la glande surrénale ; ces effets semblent liés à la pharmacologie de l'inhibition du VEGFR et/ou à l'interruption de la voie de signalisation du VEGF. Tous les effets étaient réversibles après 4 semaines sans traitement, à l'exception de ceux concernant le système squelettique (dents cassées/perdues).
Altération de la fertilité
L'étude de la fertilité et du développement embryonnaire précoce chez le rat a révélé une diminution des indices de reproduction des mâles et des femelles à des expositions d'environ 3,2 et 0,8 fois supérieures à l'ASC humaine, respectivement. Des augmentations dose-dépendantes des pertes préimplantatoires ont été observées dans la même étude.
Toxicité pour la reproduction
Une étude du développement embryo-fœtal chez le rat a révélé des effets embryotoxiques et tératogènes à des niveaux d'exposition subcliniques en l'absence de toxicité excessive chez la mère, ces effets consistant en des malformations externes, viscérales et squelettiques du fœtus. Les malformations touchaient principalement la tête, la queue, la langue, les vaisseaux sanguins, le cœur, le thymus et le squelette en développement (notamment les vertèbres).
Génotoxicité
Aucun signe de génotoxicité n'a été observé dans les études in vitro et in vivo.
Cancérogenèse
Le fruquintinib n'a pas fait l'objet d'études de carcinogénicité.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament nécessitant une surveillance particulière pendant
le traitement.
Prescription hospitalière.
Prescription réservée aux médecins compétents en
CANCEROLOGIE.
Prescription réservée aux spécialistes et services ONCOLOGIE
MEDICALE.
Gélule.
Gélule en gélatine opaque, de taille 1 (longueur approximative de 19 mm), avec une coiffe rouge et un corps blanc portant la mention « HM013 » au-dessus de « 5mg » à l'encre noire.
Flacon (45 mL) en polyéthylène haute densité (PEHD) muni d'une fermeture en polypropylène (PP) avec sécurité enfant et une cartouche dessiccante en PEHD contenant du gel de silice. La cartouche dessiccante doit être conservée à l'intérieur du flacon.
Chaque flacon contient 21 gélules. Chaque flacon est emballé dans un carton.
Chaque gélule contient 5 mg de fruquintinib.
Excipient à effet notoire
Chaque gélule de 5 mg contient 0,1829 mg de colorant rouge allura AC (E129).
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Contenu de la gélule
Amidon de maïs
Cellulose microcristalline (E460)
Talc (E553b)
Enveloppe de la gélule
Gélatine
Dioxyde de titane (E171)
Rouge allura AC (E129)
Bleu brillant FCF (E133)
Encre d'impression
Gomme laque (E904)
Propylène glycol (E1520)
Hydroxyde de potassium
Oxyde de fer noir (E172)
Processus de commande TAKEDA :
En vente directe :
01 40 67 34 34
Horaire d’ouverture : 8h30 à 17h (16h le vendredi)
Hors ouverture, week-end et jour férié : 01 40 67 32 90
Pour toute information logistique vous pouvez contacter notre service client à
l’adresse courriel suivante : France.ServiceClient@takeda.com
ou notre dépositaire Movianto pour passer vos commandes sur commande_adv@movianto.com ou par
téléphone en composant le : 01 40 67 34 34